Cold Spring Harbor, NY — In cancer cells, genetic errors wreak havoc. Misspelled genes, as well as structural variations—larger-scale rearrangements of DNA that can encompass large chunks of chromosomes—disturb carefully balanced mechanisms that have evolved to regulate cell growth. Genes that are normally silent are massively activated and mutant proteins are formed. These and other disruptions cause a plethora of problems that cause cells to grow without restraint, cancer’s most infamous hallmark.
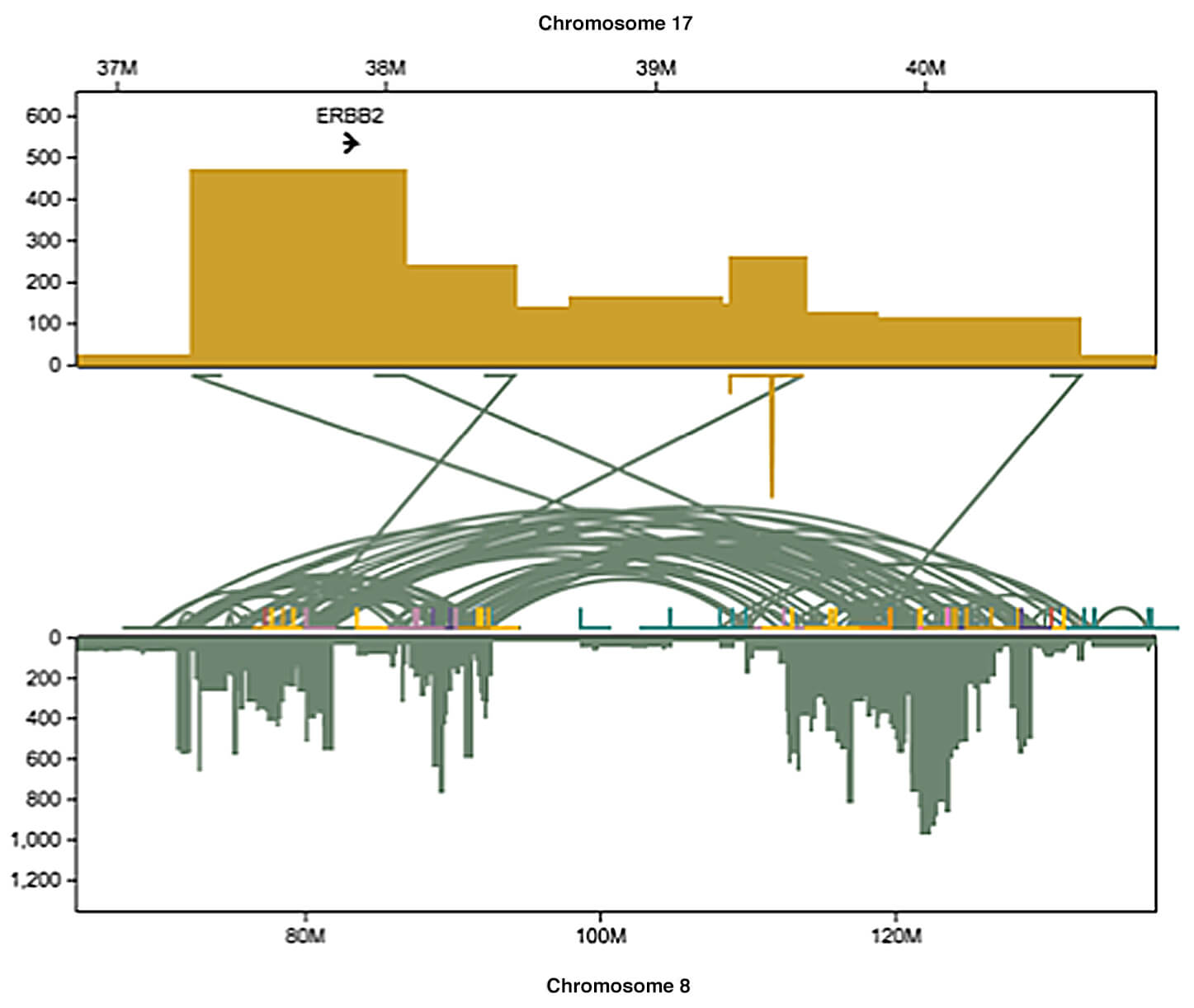
This week, scientists at Cold Spring Harbor Laboratory (CSHL) have published in Genome Research one of the most detailed maps ever made of structural variations in a cancer cell’s genome. The map reveals about 20,000 structural variations, few of which have ever been noted due to technological limitations in a long-popular method of genome sequencing.
The team, led by sequencing experts Michael C. Schatz and W. Richard McCombie, read genomes of the cancer cells with so-called long-read sequencing technology. This technology reads much lengthier segments of DNA than older short-read technology. When the results are interpreted with two sophisticated software packages recently published by the team, two advantages are evident: long-read sequencing is richer in terms of both information and context. It can, for instance, make better sense of repetitive stretches of DNA letters—which pervade the genome—in part by seeing them within a physically larger context.
The team demonstrated the power of long-read technology by using it to read the genomes of cells derived from a cell line called SK-BR-3, an important model for breast cancer cells with variations in a gene called HER2 (sometimes also called ERBB2). About 20% of breast cancers are “HER2-positive,” meaning they overproduce the HER2 protein. These cancers tend to be among the most aggressive.
“Most of the 20,000 variants we identified in this cell line were missed by short-read sequencing,” says Maria Nattestad, Ph.D., who performed the work with colleagues while still a member of the Schatz lab at CSHL and Johns Hopkins University. “Of particular interest, we found a highly complex set of DNA variations surrounding the HER2 gene.”
In their analysis, the team combined the results of long-read sequencing with results of another kind of experiment that reads the messages, or transcripts, that are being generated by activated genes. This fuller picture yielded an extraordinarily detailed account of how structural variations disrupt the genome in cancer cells and sheds light on how cancer cells rapidly evolve.
Schatz, Adjunct Associate Professor at CSHL and Bloomberg Distinguished Associate Professor at Johns Hopkins University, and McCombie, a CSHL Professor, say it is “essential to continue building a catalog of variant cancer cell types using the best available technologies. Long-read sequencing is an invaluable tool to capture the complexity of structural variations, so we expect its widespread adoption for use in research and clinical practice, especially as sequencing costs further decline.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
National Science Foundation; National Institutes of Health; CSHL Cancer Center; Watson School of Biological Sciences; Pacific Biosciences. Two employees of Pacific Biosciences were involved in the research and co-authored the paper. The SKBR3 genome was assembled with the support of DNAnexus.
Citation
Nattestad M et al, “Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line” is available online ahead of print and will appear in the August 2018 issue of Genome Research.
